| IMIM |
|
UPF |
|
CRG |
|
GRIB |
|
DATASETS |
|
Splice Site Identification by idlBNs |
| Genome BioInformatics Research Lab |
| Help | News People Research | Software | Publications | Links |
| Resources & Datasets | Gene Predictions | Seminars & Courses |
|
|
R. Castelo* and R. Guigó Bioinformatics, 20(1):i69-i76, 2004 [full text] *To whom correspondence should be adressed.
In this site we provide supplementary information consisting of the datasets specifically
built for this work as well as the full set of results.
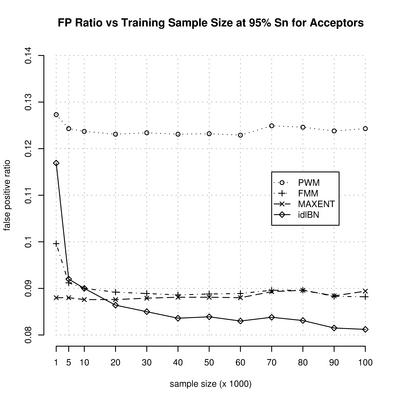
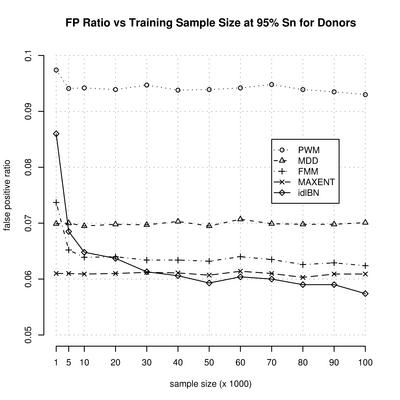
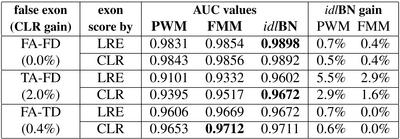
Motivation: Computational identification of functional sites in nucleotide sequences is at the core of many algorithms for the analysis of genomic data. This identification is based on the statistical parameters estimated from a training set. Often, because of the huge number of parameters, it is difficult to obtain consistent estimators. To simplify the estimation problem, one imposes independence assumptions between the nucleotides along the site. However, this can potentially limit the minimum value of the estimation error. Results: In this paper we introduce a novel method, in the context of identifying functional sites, that finds a reasonable set of independence assumptions supported by the data, among the nucleotides, and uses it to perform the identification of the sites by their likelihood ratio. More importantly, in many practical situations it is able to improve its performance as the training sample size increases. We apply the method to the identification of splice sites, and further evaluate its effect within the context of exon, and gene, prediction.
In the experiments we have used three different datasets. One is the set of 19,174 human annotations in the reference sequence (RefSeq) dataset (UCSC version hg15) based on NCBI Build 33 (April 10, 2003). The other two are used exclusively for testing purposes and correspond to the Burset and Guigó (1996) dataset of 570 human genes (BG-570), and the Rogic et al. (2001) dataset of 195 genes (HMR-195) including human (103), mouse (82) and rat (10) genes.
These datasets have been further transformed to obtain the training and testing
datasets of acceptors and donors (ACCDON), the training datasets of start, stop, acceptor
and donor sites (NOBGRORS), and the testing datasets of internal exons from the BG-570
and HMR-195 genes (BGROIEXONS). Below we can find the detailed description of each of
them as well as the corresponding data files.
ACCDON datasets In order to assess splice site identification we have extracted from the
RefSeq genes two sets of non-redundant canonical donor and acceptor sites. The
non-redundancy has been enforced by selecting unique stretches of DNA
containing the donor and the acceptor site, where the donor stretch had 39bp
(the first 3bp in the exon) and the acceptor stretch had 23bp. Afterwards, from
the donor context we selected 9bp (3bp exon + gt + 4bp intron) to form every
donor site. This left a total of 124,727 donor sites, and 130,220 acceptor
sites. We created 2 datasets of these same sizes with false (decoy) donor and
acceptor sites by sampling uniformly from coding regions, and 2 more datasets
by sampling from intronic regions of the RefSeq genes. All the false sites
matched the corresponding minimum consensus (gt,ag). Non-redundancy was
enforced in the same way as for the true sites. Below we can find the corresponding
files:
The donor and acceptor site prediction has been assesed by carrying out a 10-fold cross-validation along increasing sizes of the training datasets of donor and acceptor sites, starting on 1,000, 5,000 and 10,000, and continuing with sizes that increase in 10,000 sites up to 100,000. The rest of the sites not contained in each training dataset form the test dataset. The 10-fold cross-validation is done by sampling each of the 12 sizes, ten times from the ACCDON datasets, where half of the false sites were sampled from the file with those from coding regions and half from the file with those from intronic ones. This led to 12 x 10 x 4 = 480 datasets that have a total compressed size of about 780Mbytes. Because of such a large size, we do not include the link here, but we can provide the tar ball on request. NOBGRORS datasets From the RefSeq data, we have filtered out genes with either the same name, or annotated in a loose chromosome chunk (chr_random), or those that overlap another annotation (leaving one copy arbitrarily chosen). From this latter set, we translated into protein each gene, and discard those that either did not start, and end, with a start, and stop, codons, and those which had in-frame stop codons. This left a total of 14,080 genes. Further, we obtained the corresponding proteins from the BG-570 and HMR-175
genes and performed BLASTP against the previously filtered
RefSeq proteins. These are the files involved:
From the previous BLASTP output, we considered only those HSPs that had an E-value smaller
or equal than 10-5 and from these, select those that showed an identity
greater than or equal to 60%. There were 855 proteins matching these
criteria, and the corresponding genes were removed from the RefSeq genes,
leaving a total of 13,225 genes, which form the NOBGRORS dataset from which we extracted
the following sets of functional (real and false) sites:
All the false sites are formed by sampling half of them from coding regions and half of them from intronic regions. The entire set of 13,225 genes has a total compressed size of 177Mbytes, for which we do not include a link here but we can provide on request. BGROIEXONS datasets From the BG-570 and HMR-195 genes datasets, we extracted all the internal exons (2,110).
From each gene with n internal exons, we have randomly sampled the same amount
of false exons three times. First, ensuring that both splice sites are false;
second, for every true acceptor site, we chose randomly a downstream false
donor site; and third, for every true donor site we chose randomly an upstream
false acceptor site. These are the resulting datasets:
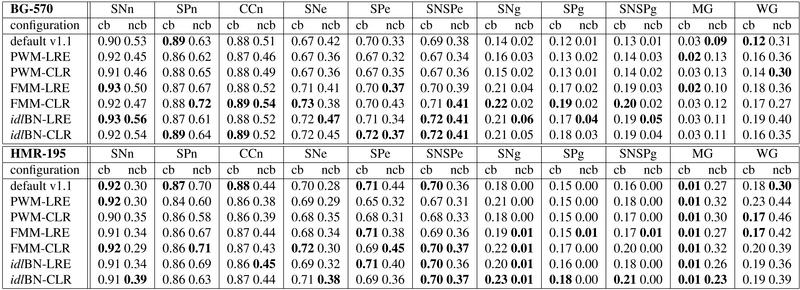
We include here the full set of results on splice site, internal exon, and gene, prediction.
Splice site prediction
Internal exon prediction
Gene prediction
We denote by inclusion-driven learned Bayesian networks (idlBNs) those
Bayesian networks whose structure is learned by an inclusion-driven algorithm.
We have used the HCMC algorithm, introduced by
Castelo and Kocka (2003).
|
|
||||||